| Titel: | Die Raoult'sche Methode der Molekulargewichtsbestimmung; von Constantin Klinge. |
| Autor: | Constantin Klinge |
| Fundstelle: | Band 273, Jahrgang 1889, S. 218 |
| Download: | XML |
Die Raoult'sche Methode der
Molekulargewichtsbestimmung; von Constantin Klinge.
(Fortsetzung der Abhandlung S. 179 d.
Bd.)
Mit Abbildungen auf Tafel
11.
Die Raoult'sche Methode der
Molekulargewichtsbestimmung.
A. F. HollemannBerichte, XXI, 860. hat ein
noch einfacheres Verfahren in Anwendung gebracht.
Das Gefäſs, worin sich die auf ihren Gefrierpunkt zu untersuchende Flüssigkeit
befindet, ist ein weites Probirrohr (etwa 2cm
Durchmesser); es wird durch die Klemmschraube eines Stativs festgehalten. Im
Probirrohre hängt ein in 1/10 Grade getheiltes, empfindliches Thermometer;
weiter ist noch ein Rührer (ein am unteren Ende umgebogener Glasstab) darin
befindlich. Als Kühlgefäſs wird ein mit Eiswasser gefülltes Becherglas benutzt, das
am selben Stativ auf einem mit Drahtnetz versehenen Ring steht und während des
Versuches auf und ab gehoben wird, wogegen die relative Lage von Probirrohr und
Thermometer unverändert bleiben.
Bei Ausführung eines Versuches kühlt man die zu untersuchende Flüssigkeit (wovon 30
bis 40g ausreichen) ungefähr ab bis 0,5° unter den
Gefrierpunkt des Lösungsmittels; der Rührer wird dabei mit der Hand in Bewegung
gehalten. Danach wird das Becherglas mit Eiswasser ganz vom Probirrohre weggenommen.
Durch Reiben mit dem Rührer an der Glaswand, oder sicherer durch Einbringen eines
minimalen Krystallflitterchens wird jetzt die Krystallisation eingeleitet. Sobald
diese eintritt, sieht man die Temperatur, die bis dahin noch stets sinkend geblieben
ist, plötzlich steigen. Man wartet einige Augenblicke, rührt die Flüssigkeit nun um
und liest die Temperatur ab mit einer kleinen Wollaston'schen Lupe, wie sie auch sonst im Laboratorium oft benutzt wird.
Dies wird in kurzen Intervallen noch zwei- bis dreimal wiederholt, vor jeder
Ablesung erst gerührt, um sich zu überzeugen, daſs Constanz der Temperatur
eingetreten ist.
Man thaut jetzt die Kryställchen wieder auf, das Probirrohr mit der Hand oder mit ein
wenig lauwarmem Wasser erwärmend, und wiederholt dann in derselben Weise die
Gefrierpunktsbestimmung noch zweimal. Die drei so erhaltenen Gefrierpunktszahlen
differiren dann höchstens um 2/100 Grad.
Als Beweis, daſs dieses höchst einfache Verfahren für den Zweck ausreicht, gibt Hollemann die folgenden Molekulargewichtsbestimmungen,
die danach ausgeführt worden sind, an:
Proc. Geh.der Lösung
Gefrierpunkts-Erniedrigung
A(Mittel)
Mol.-Gw.Gefunden
Mol.-Gw.Berechnet
1) Benzamid
1,96
0,62; 0,61; 0,61
0,31
126
121
2) Phtalsäureanhydrid
1,57
0,35; 0,35; 0,35
0,23
169
148
3) Acetophenon
1,82
0,55; 0,55; 0,55
0,30
130
120
4) Naphtalin
1,87
0,54; 0,55; 0,55
0,29
134
128
Die Ausführung einer Molekulargewichtsbestimmung nach diesem hier beschriebenen
Verfahren dürfte, das Herstellen der Lösung, wie auch die Gefrierpunktsbestimmung
des Lösungsmittels selber mitgerechnet, kaum mehr als ¾ Stunden in Anspruch
nehmen.
Auf Veranlassung des Herrn Prof. Engler habe ich mich
längere Zeit mit der Raoult'schen Methode beschäftigt.
Durch die von mir gemachten Beobachtungen wurden schlieſslich die beiden eben
angegebenen Verfahren gewissermaſsen mit einander vereinigt und die Versuche in
folgender Weise angestellt:
Der Mantel zur Aufnahme der Flüssigkeit erhielt die Gröſse, daſs 50g Lösungsmittel denselben ungefähr bis zur Hälfte
füllten.
Zum Schütze gegen die Feuchtigkeit der Luft wurde der Mantel mit einem doppelt
durchbohrten Kork verschlossen. In die eine Bohrung wurde ein in zehntel Grade
getheiltes Thermometer, in die andere ein Rührwerk, welches genau in der von Auwers angegebenen Weise ausgeführt war, gesteckt. Der
ganze Apparat wurde in ein groſses Becherglas mit Wasser gesenkt, dessen Temperatur
sich etwa 2° unter der jedesmaligen Erstarrungstemperatur des Lösungsmittels befand,
und die Versuche unter Einhaltung der von Auwers
angegebenen Vorschriften angestellt. Nur habe ich den Krystalleinwurf ganz
weggelassen, da beim Abkühlen des Lösungsmittels (Eisessig oder Phenol) auf etwa
0,5° unter seinen Erstarrungspunkt die Erstarrung von selbst vor sich geht und man
ganz normale Werthe erhält. – Auf diese Weise wurde erreicht, daſs zwischen den drei
Gefrierpunktszahlen fast niemals sich eine Temperaturdifferenz ergab, so daſs in der
Folge schon der erste beobachtete Werth als brauchbar angenommen werden konnte. Ich
neige mich daher zu der Ansicht, daſs diese Differenzen bei Auwers durch den Krystalleinwurf, bei Hollemann dagegen durch den Umstand, daſs ein offenes Gefäſs angewandt
wird, hervorgerufen werden.
Für meine Resultate mag das folgende Versuchsbeispiel sprechen:
Triphenylpyridin, C23H17N, M = 307.
Erstarrungspunkt des Phenols: 39,00°.
Angewandt: 0g,3235 Triphenylpyridin in 52g,0 Eisessig.
Gefunden:
E
C
A
M
33,84°
0,16°
0,257°
311
33,83°
0,17°
0,273°
296
33,83°
0,17°
0,273°
296
–––––
––––
Mittel
0,268°
300.
Erstarrungspunkt des Phenols: 39,10°.
Angewandt: 0g,2050 Triphenylpyridin in 52g,8 Eisessig.
Gefunden:
E
C
A
M
38,960°
0,140°
0,259°
309
38,960°
0,140°
0,259°
309
38,960°
0,140°
0,259°
309
–––––
––––
Mittel
0,259°
309
Theorie
Mittel der Versuche
M = 307
M= 304,5.
Handelt es sich um die Bestimmung des Molekulargewichtes einer Substanz in einem
Lösungsmittel, dessen molekulare Depression T schon
bekannt ist, so wird man bei Anwendung eines der drei eben beschriebenen Verfahren
in den meisten Fällen befriedigende Werthe erhalten.
Hentschell, welcher Versuche über das gegenseitige
Verhalten von Benzol und Eisessig angestellt und zu seinen Bestimmungen
ausschlieſslich Substanzen von flüssigem Aggregatzustande verwendet hat, benutzt
einen ApparatZeitschr. für phys. Chem., II, 306.
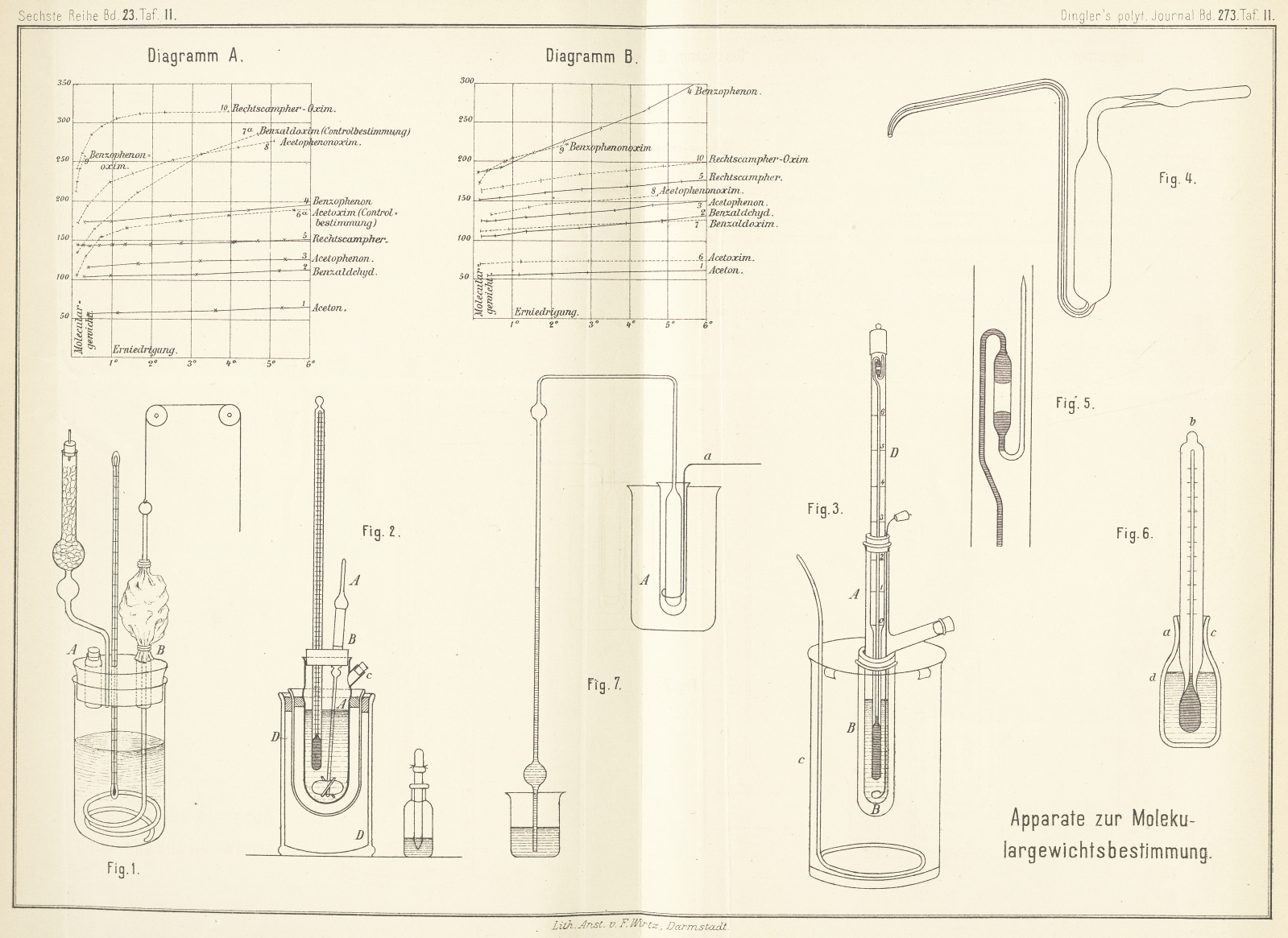
(Fig. 2),
der von dem Auwers'schen abweicht.
Zu genaueren Bestimmungen ist es, zumal wenn man das leichtflüchtige Benzol als
Lösungsmittel benutzt, unbedingt nöthig, im abgeschlossenen Raume zu arbeiten,
namentlich, wenn man durch Ausführung einer Reihe von Bestimmungen das Verfahren in
die Länge zieht. Die Bewegung der theilweise erstarrten Flüssigkeit wird durch das
Wirbeln eines an der Glasbläserlampe hergestellten Flügelrades erzielt, dessen Stiel
A den Stöpsel des Versuchsgefäſses durchsetzt,
wobei durch ein eingeschobenes Glasröhrchen für leichte Führung gesorgt ist; diese
Achse des Flügelrades steht etwas schief, so daſs das flügeltragende Ende des
Stieles genau im Mittelpunkte der Gefäſskuppel steht. Die drehende Bewegung des
Flügelrades wird dadurch bewirkt, daſs man leise an dem aufgekitteten Glasrohre B entlang fährt; ist alles sorgfältig eingerichtet, so
genügt diese Bewegung, um den Inhalt des Gefäſses heftig durch einander zu
wirbeln.
Da es sich meist um Reihen von Bestimmungen handelt, so wird die zu untersuchende Flüssigkeit
in ein mit eingeschliffenem Tropfrohre versehenes Fläschchen gethan, welches nach
jedesmaligem Eintragen von Flüssigkeit zurückgewogen wird.
Das Eintragen geschieht durch den mit Kork verschlossenen Stutzen C. Die Beobachtung des Erstarrungspunktes kann in
zweierlei Weise geschehen. Man läſst entweder die Lösung erstarren und beobachtet
unter beständigem Umrühren den Wärmegrad des Thermometers, bei welchem eine eben
noch sichtbare Wolke von Krystallen übrig geblieben ist, oder man läſst vor dem
Eintragen der zu untersuchenden Flüssigkeit einen Theil des Lösungsmittels oder der
bereits gewonnenen Lösung erstarren, um nun erst von jener Flüssigkeit zuzutropfen;
bei gleichmäſsigem Rühren sinkt die Temperatur jetzt sehr rasch und stellt sich um
so genauer auf den Erstarrungspunkt ein, je zarter der Flor von Krystallen ist,
welcher nach Zusatz der Versuchsflüssigkeit dem Verthauen widerstanden hat.
Natürlich ist der Versuch miſsglückt, wenn alle Krystalle nach dem Eintragen
verschwinden, und wird dann die Bestimmung des Schmelzpunktes nach dem zuerst
angeführten Verfahren nachgeholt. Das zweite Verfahren eignet sich besonders bei
Benutzung von Eisessig als Lösungsmittel; das Verfahren beruht auf dem
auſserordentlichen Ueberwiegen der latenten Schmelzwärme gegenüber der specifischen
Wärme.
Bei Bestimmung der Schmelzpunkte der Lösungsmittel selbst thut man gut, dieselben
vorsichtig überkalten zu lassen, worauf sie in ihrer ganzen Masse in kleinen,
leichtlöslichen Krystallen erstarren, anderenfalls scheiden sich leicht Krusten an
den Wänden des Gefäſses ab, welche genaue Bestimmungen unmöglich machen; mit Zunahme
des gelösten Körpers hört diese Krustenbildung auf.
Man hält zweckmäſsig doppelwandige Standgefäſse (vgl. DD
auf Fig. 2)
bereit, welche man trocken als Schutzmittel zur Abhaltung von warmer Zimmerluft
oder, mit Eiswasser gefüllt, zur Kühlung der Lösungen benutzt.
Bezüglich der Resultate sei auf die OriginalarbeitZeitschr. für phys. Chem., II, 308.
verwiesen.
Für sehr genaue Untersuchungen, z.B. bei Bestimmung der molekularen Depression eines
Lösungsmittels, leistet ein von BeckmannZeitschr. für phys. Chem., II,
638. construirter Apparat (Fig. 3) vortreffliche
Dienste. Das Gefäſs A, welches die zu prüfende
Flüssigkeit aufnimmt, besteht aus einem starkwandigen groſsen Probirrohre, welches
seitlich einen Stutzen trägt, behufs Einfüllung der Substanz. Um eine Bestimmung
auszuführen, gibt man in das zuvor mit einigen scharfkantigen Platinschnitzeln
beschickte und tarirte Probirrohr, welches bis zum Stutzen etwa 25cc faſst, ungefähr 15g Lösungsmittel, trocknet den oberen Theil des Rohres mittels
Filtrirpapier und wägt nun bis auf Centigramme genau. Nachdem der aus dickem Platindrahte
bestehende Rührer eingelassen ist, wird das Thermometer mittels Kork aufgesetzt. Um
das Probirrohr befestigt man zunächst mit Kork einen weiteren Cylinder B, der als Luftmantel dient, erst dieser wird in das
Batterieglas C eingesenkt, welches mit Kühlflüssigkeit
gefüllt ist.
Zweckmäſsig hält man die Temperatur in dem Batterieglase etwa 2 bis 5° unter dem
Erstarrungspunkte der zu prüfenden Flüssigkeit. Bei Arbeiten mit Eisessig, dessen
Schmelzpunkt bei rund 16° liegt, läſst sich eine zu hohe Temperatur durch Einwerfen
von Eisstücken und Umrühren mit dem äuſseren Rührer herabdrücken. Ohne Luftmantel
wäre das natürlich während der Arbeit nicht statthaft. Wird Benzol, welches bei rund
5,5° schmilzt, verwendet, so füllt man das äuſsere Gefäſs zum groſsen Theile mit
Eisstücken und läſst es dann voll Wasser laufen. Die Sorge um die äuſsere Temperatur
fällt hier bei genügend vorhandenem Eise fort, bis der Gefrierpunkt der zu prüfenden
Lösung unter 2° sinkt. Wird stärkere Abkühlung nothwendig, wie es bei Anwendung von
Wasser als Lösungsmittel von vornherein der Fall ist, so gibt man zu der Mischung
von Eis und Wasser im äuſseren Gefäſse unter Umrühren so viel Kochsalz, bis die
gewünschte Temperatur erreicht ist. Ein beständiges Sichtbarbleiben des
Gefriergefäſses ist ganz überflüssig. Nach einiger Uebung braucht man die äuſsere
Temperatur gar nicht mehr mit dem Thermometer zu controliren; die Schnelligkeit, mit
welcher die Temperatur im inneren Gefäſse sinkt, genügt zur Beurtheilung.
Nach dem Abkühlen der Flüssigkeit unter ihren Gefrierpunkt wird für den Beginn der
Krystallabscheidung Sorge getragen und das bei beständigem Rühren nun rasch
steigende Quecksilber des Thermometers gibt in seinem höchsten Stande den
Gefrierpunkt an. Auch bei diesem Verfahren wird das Einwerfen von Krystallen, um die
Erstarrung einzuleiten, weggelassen. Um die Möglichkeit einer Abkühlung des
Lösungsmittels zu beschränken, ist das Probirrohr mit Platinschnitzeln beschickt und
mit einem auf und ab gehenden, Erschütterungen erzeugenden Rührer versehen worden.
Bei Anwendung von Benzol hat dies den Erfolg, daſs der Quecksilberfaden nur wenige
Hundertstel-Grade unter den Gefrierpunkt sinkt, um sich in Folge einer geringen
feinpulverigen Krystallabscheidung alsbald sehr genau auf den Gefrierpunkt
einzustellen.
Eisessig läſst sich unter diesen Bedingungen etwas stärker, bis zu 0,5°, Wasser bis
zu 1° überkühlen. Für die Bestimmung des Gefrierpunktes der reinen Lösungsmittel ist
die in den letzteren beiden Fällen auftretende stärkere Eisabscheidung ohne Belang;
wie für concentrirtere Lösungen der entstehende Fehler leicht vermieden wird, soll
sogleich erörtert werden.
Nachdem der Gefrierpunkt des Lösungsmittels auf diese Weise bestimmt und nach Aufthauen des
abgeschiedenen Eises durch wiederholte Bestimmung auf seine Constanz geprüft worden
ist, wird die zu untersuchende Substanz durch den Stutzen eingeführt und nach
erfolgter Lösung – dem Stutzen anhaftende Partikeln können durch Neigen weggespült
werden – der Gefrierpunkt aufs Neue zweimal bestimmt. Durch Subtraction erfährt man
ohne Weiteres die stattgehabte Erniedrigung. Nach Zufügung einer weiteren Menge
Substanz kann sofort die Bestimmung für höhere Concentrationen angeschlossen werden.
Bei der Untersuchung von Lösungen tritt mit steigender Concentration immer mehr die
Nothwendigkeit hervor, eine stärkere Ueberkühlung möglichst zu vermeiden, d.h. die
Menge des ausfrierenden Lösungsmittels thunlichst zu beschränken. Da nur dieses sich
ausscheidet, muſs mit dessen Entfernung die zurückbleibende Lösung concentrirter
werden und einen immer niedrigeren Schmelzpunkt zeigen. Die möglichen Fehler werden
bei obigem Verfahren um so gröſser, wenn, wie das besonders bei Eisessig und Wasser
der Fall ist, durch die gelöste Substanz die Krystallabscheidung in höherem Maſse,
unter Umständen um viele Grade, hintangehalten wird. Aber auch in diesen Fällen kann
man ohne Einbringen von fertigem Eise einen hohen Grad von Genauigkeit erreichen.
Nachdem Eisausscheidung durch Abkühlung ohne Luftmantel bei kräftigem Umrühren
hervorgerufen ist, läſst man während kurzer Ruhe am Boden des Gefriergefäſses eine
ganz dünne Schicht des Lösungsmittels anfrieren, thaut sodann die in der Flüssigkeit
schwebende feinzertheilte Abscheidung, welche viel leichter zergeht als die dünne
Eiskruste, fast völlig auf, sistirt weitere Erwärmung durch Einsetzen in Luftmantel
und Kühlflüssigkeit und führt, wenn das Thermometer zu sinken beginnt, die
Bestimmung wie früher aus. Durch einige Uebung gelingt es leicht, den Versuch so zu
leiten, daſs bei einer Ueberkühlung von 0,1° und weniger Graden bereits genügend
feinzertheiltes Eis ausgeschieden ist, um das Thermometer wieder ansteigen zu
lassen.
Zur Vermeidung grober Täuschungen verlasse man sich bei diesen Versuchen nie allein
auf den Gang des Quecksilberfadens, sondern betrachte die Beobachtung nicht eher als
sicher, bis man Sich von der wirklich erfolgten Abscheidung fein zertheilten Eises
überzeugt hat.
Zum Einbringen fester Substanz in den Apparat dient ein einseitig zugeschmolzenes
Glasrohr von einem Durchmesser, daſs es bequem durch den Stutzen geht. Für die
Einführung von Flüssigkeiten empfiehlt sich überaus der nachstehend abgebildete
(Fig. 4),
leicht aus Glas herzustellende Apparat, welcher nur eine Modification des Sprengel-Ostwald'schen PyknometersJ. f. pr. Chem., [2] 16, 396.
darstellt. Der Apparat wird gefüllt, indem man die Kapillare, welche unten am
cylindrischen Gefäſse angeschmolzen ist, in die Flüssigkeit eintaucht, das obere Knierohr zum
Schütze gegen Feuchtigkeit mit einem Chlorcalciumrohre verbindet und nun ansaugt.
Die Entnahme von Substanz geschieht durch Einblasen, während die Kapillare in den
Stutzen geschoben ist. Eines vollkommenen Abtropfens halber ist die Kapillare an der
Mündung abwärts gebogen und schief angeschliffen.
Auch sehr leicht flüchtige Flüssigkeiten können vor einem Verdunsten bewahrt werden,
wenn man die Kapillare recht eng nimmt und das obere Rohr, wie in der Figur, an
einer Stelle kapillar auszieht.
Was aber den Apparat besonders vor den vorhergehenden auszeichnet, ist das
empfindliche, von Beckmann eigens für den Apparat
construirte Thermometer (Fig. 3
D), welches durch Billigkeit, Handlichkeit,
Zuverlässigkeit und Anwendbarkeit bei allen hier in Betracht kommenden Temperaturen
von – 6° bis + 60° ausgezeichnet ist.Der Glastechniker F. O. R. Goetze in Leipzig
liefert dieses Thermometer aus Jenaschem Normalglase zum Preise von 25 M.
Derselbe fertigt auch die obigen Apparate, welche übrigens mit den
Hilfsmitteln eines jeden Laboratoriums leicht hergestellt werden
können.
(Schluſs folgt.)
Tafeln